Research Areas
The Jain Laboratory was part of the University of California, San Francisco for 22 years (1999-2021). It is now Jainlab LLC, established to maintain and expand the academically focused distribution of data relating to the following research areas:
- Small-molecule conformer generation, including complex macrocycles
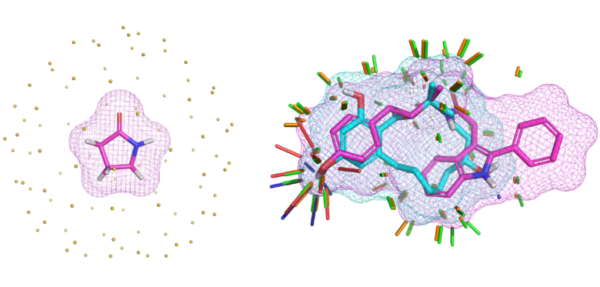
- Molecular docking and protein binding site comparison and analysis
- Three-dimensional molecular similarity and multiple ligand alignment
- Prediction of affinity and pose for biologically interesting molecules
- Improvements to modeling X-ray density, particularly by employing conformational ensembles
The work represents a unique approach in CADD by respecting physics, chemistry, and biology while also taking machine learning and parameter optimization seriously.
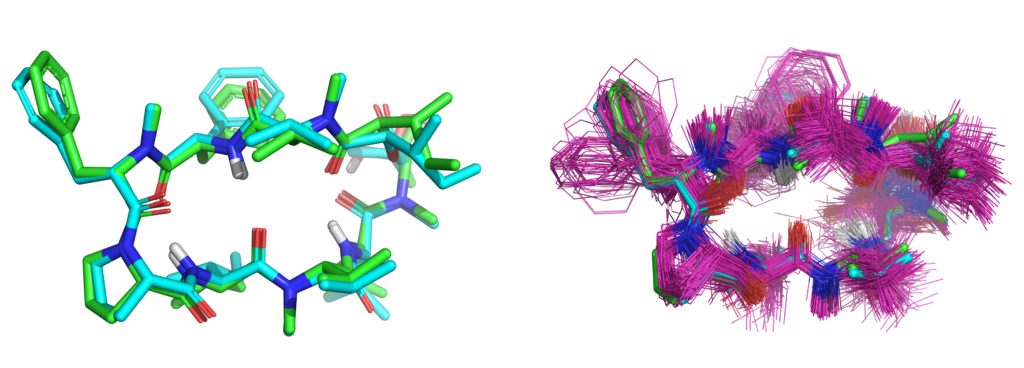


What’s New?
JMC: Bound Ligand Strain
January 27, 2023
We have published a comprehensive study of bound ligand strain in J. Med. Chem., with our colleagues from Merck. Ligand strain, when properly estimated using real-space refinement, is generally quite low. But it grows more rapidly than ligand size and follows a simple distributional model. The super-linearity of ligand strain w/r/t size explains a part of the challenge in designing larger ligands that are highly efficient.
JCAMD: Macrocycle Optimization
August 3, 2023
We have published, a comprehensive study of complex macrocycle optimization through careful application of NMR restraints combined with molecular docking, molecular similarity, and estimation of bound ligand strain with colleagues from BMS.
JMC: xGen Papers
March 17, 2021
We have published a second paper with colleagues from Merck looking at peptide macrocycle strain energetics in the context of xGen refined conformational ensembles. This follows the paper entitled “XGen: Real-Space Fitting of Complex Ligand Conformational Ensembles to X‐ray Electron Density Maps.” We show that conformational ensembles, without atom-specific B-factors, are better models for ligands in terms of both fit to X-ray density and strain energy.
QuanSA Affinity Prediction Method
February 26, 2024
A paper describing an active-learning approach to lead optimization of a macrocyclic fungicide natural product has been accepted for publication in JCAMD and will appear shortly. This builds upon two prior papers: 1) a 2021 paper shows QuanSA to be complementary and synergistic with predictions from FEP+ across 17 targets; and 2) the introductory paper offers additional details about the QuanSA multiple-instance machine-learning method for binding affinity and pose prediction.